Medical Tripos 1A
The Basics
The 3 years of your undergraduate at Cambridge can be divided up into parts 1A (first year), 1B (second year), and part II.
1A encompasses 3 major subjects, Functional Architecture of the Body (FAB), a.k.a Anatomy, Molecules in Medical Science (MIMS), aka Biochemistry, and Homeostasis (HOM), which is physiology.
FAB
- You’ll have 2 dissections in one week, and then 1 dissection the following week
- There will be lectures that run in parallel, these can also be tested in MCQs
- I’d recommend memorising the parts of the manual corresponding to each session before the sessions -> this really helps you to get the most out of dissection
- Course Breakdown
- Upper Limb – really make sure you know the origins and attachments of the muscles, this is very valuable for the MCQs
- Thorax – probably the easiest topic there is, very easy to visualise
- Abdomen and Pelvis – the hardest topic to visualise, it’s worth putting in the work to make sure you understand this during term, because it’ll be a pain to go over it in the holidays
- Lower Limb – same advice as for upper limb, this topic is probably the smallest one
- Embryology – these lectures run in parallel with what is covered during dissection, and also can come out in the MCQs
MIMS
- 3 lectures a week, 1 practical a term (with a discussion session 2 weeks later), 2 PBL presentations across Michaelmas and Lent
- Michaelmas
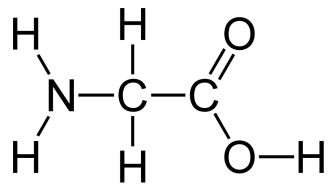
- You’ll start off with an introduction to diabetes, with 5 lectures on protein structure and enzyme activity
- After this, the more nitty gritty stuff comes in, in the form of metabolism
- This carries on for a while, the term finally ends off with cell signalling and protein sorting, the former is very useful for HOM, so I’d make an effort to try to learn this during term
- Lent
- The major theme of this term is molecular biology and the clinical linker is cancer, so the prologue and epilogue lectures will be centered on this
- You’ll start off with the basics of the genome, then gene expression, followed by broad genetics
- The term ends off with the cell cycle and cell death
- There are also 2 lectures just on cancer; you can build on these for essays to score.
- There is a lot of very specific detail you are expected to know, so build a system over the Christmas vacation that lets you learn it efficiently
- The practical paper is quite challenging, and doesn’t really get much attention in the practical sessions themselves, so I would recommend practising these over the Easter vacation
- Make a list of experimental techniques so when you’re asked to suggest further experiments, you can jot down some good ideas when asked in the practical paper
HOM
- 3 lectures a week
- HOM doesn’t require as much brute force memorisation as MIMS or FAB, but you do have to make an effort to wrap your head around the logic of certain concepts.
- These are the major topics with some advice for each
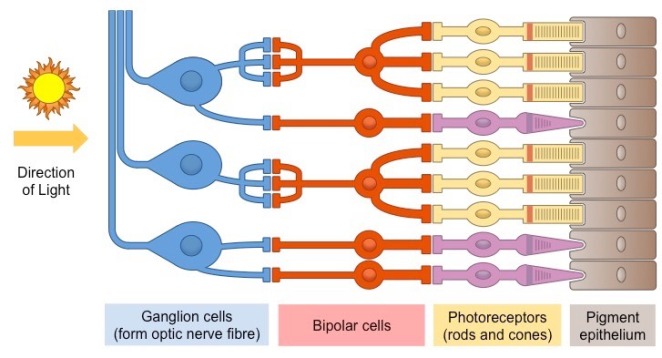
- Nerves
- Quite a mathematical topic, you can usually relate most concepts to the Nernst equation, which I’d recommend understanding the derivation of
- Learn the experiments; they could test them in MCQs, or you could use them in essays
- Supervisions should give you some additional diagrams to use in essays, the ones brought up in lectures are a bit basic
- Muscles
- Create a table comparing the different kinds of muscles, it helps to produce a nice mental schema for compare and contrast essay questions
- Cardiovascular
- Very content heavy topic, but well taught
- Make sure you understand Starling’s Law, because a good understanding of it helps for both respiratory and renal
- Respiratory
- Get your head around what increases/decreases compliance early on, it makes life much easier later
- There are a lot of numbers to know for this topic, so make sure you use the right units for them
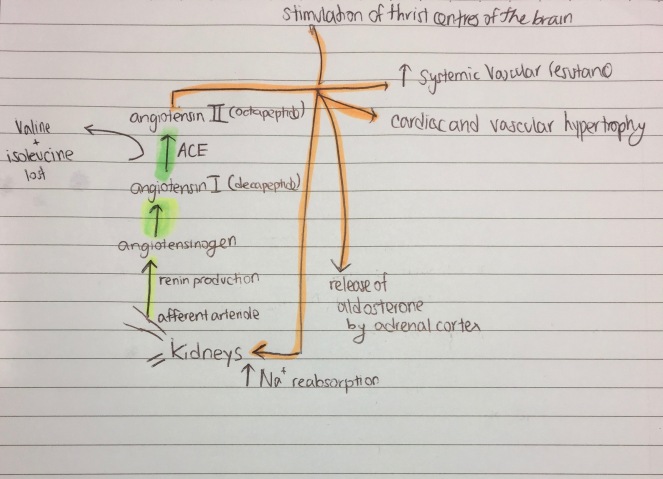
- Renal
- Again, very content heavy, and it’s the longest lecture series
- Make a table for all the transporters, and for hyper and hypokalaemia/calcaemia
- Get the Koeppen monograph – it explains stuff clearly and has loads of extra stuff you can use in essays
- Digestion
- One of the easiest HOM topics, but the most content per unit lecture
- Create a table for the GI hormones
- Endocrine
- This is actually a pretty good lecture series, probably worth going for, even though it’s in Easter
Some of My Resources
FAB
- A set of tables I made summarising the anatomy of the upper limb, with blanked out tables by the side for testing
MIMS
HOM
Have fun!